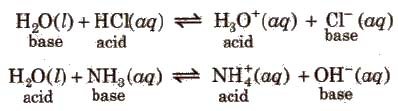Gli alogeni sono elementi non metallici appartenenti al Gruppo 17 della Tavola Periodica dotati di elevata reattività. Gli elementi costituenti questo gruppo sono: fluoro, cloro, bromo, iodio, astato e Tennessine.
Il termine alogeno fu proposto dal chimico svedese Jöns Jacob Berzelius nel 1842 deriva dal greco –ἅλς e γεν- che significa formatore di sali.
E’ l’unico Gruppo della Tavola Periodica in cui sono presenti elementi allo stato gassoso (fluoro e cloro), liquido(bromo) e solido (iodio, astato e tennessine).
La chimica degli alogeni è limitata ai primi 4 elementi del gruppo stante la radioattività degli ultimi due che hanno marcata radioattività e tempi di dimezzamento molto bassi.
Gli alogeni, allo stato elementare formano molecole biatomiche di tipo covalente puro in cui gli elementi sono legati tra loro tramite un legame semplice, mentre nei sali gli alogeni si trovano come ioni alogenuro F–, Cl–, Br–, I–.
Gli ioni fluoruro vengono rinvenuti in natura nei minerali fluorite sotto forma di CaF2 e criolite Na3AlF6, gli ioni cloruro nei minerali halite sotto forma di NaCl, silvite sotto forma di KCl e carnallite sotto forma di KMgCl3·6(H2O) nonché nei mari e ni oceani. Gli ioni bromuro e ioduro sono presenti in piccola quantità nell’acqua di mare.
Scendendo dall’alto verso il basso lungo il gruppo il colore delle molecole biatomiche diviene via via più scuro: il fluoro è praticamente incolore, il cloro è giallo-verde, il bromo rosso-marrone e i cristalli di iodio viola scuro.
L’energia di ionizzazione, l’elettronegatività e la potere ossidante diminuisce scendendo dall’alto verso il basso lungo il gruppo mentre il potere riducente degli alogenuri diminuisce dal basso verso l’alto.
Potere ossidante: F2 > Cl2 > Br2 > I2
Potere riducente: I– > Br– > Cl– > F–
Gli alogeni possono essere ottenuti facendo reagire una soluzione contenente un alogenuro con un alogeno avente maggiore potere ossidante; ad esempio:
2 I– + Br2 → I2 + 2 Br–
Il bromo infatti fu ottenuto dal chimico francese Antoine Jérôme Balard nel 1826 facendo reagire ioni bromuro con una soluzione contenente Cl2 secondo la reazione:
2 Br– + Cl2 → Br2 + 2 Cl–
Per ottenere il cloro è tuttavia necessario un agente ossidante particolarmente forte come il biossido di manganese:
2 Cl– + MnO2 + 4 H+ → Cl2 + Mn2+ + 2 H2O
Il metodo migliore per ottenere il cloro gassoso è comunque quello elettrolitico; il cloro può essere ottenuto dall’elettrolisi del cloruro di sodio fuso:
2 NaCl → 2 Na + Cl2
La sintesi del fluoro avvenne molto più tardi sia per la difficoltà di trovare un ossidante sufficientemente forte da far avvenire la semireazione di ossidazione 2 F– → F2 sia per il fatto che sia il fluoro che il fluoruro di idrogeno sono molto tossici. Fu solo nel 1886 che il chimico francese Francis Ferdinand Moissan mise a punto un metodo industriale per l’ottenimento del fluoro per elettrolisi di una miscela di KHF2:
2 KHF2 → H2 + F2 + 2 KF
Gli tutti gli alogeni formano acidi binari forti ad eccezione dell’acido fluoridrico che è un acido debole; il fluoro, che è l’elemento più elettronegativo ha sempre e solo numero di ossidazione -1, mentre gli altri elementi presentano numeri di ossidazione -1, +1, +3, +5 e +7 in quanto il fluoro non può avere un’espansione della sfera di valenza al contrario degli altri alogeni che, avendo orbitali d vuoti possono dare tale tipo di espansione.
Esempi di composti degli alogeni con i diversi numeri di ossidazione sono riportati in tabella:
| Composto |
Numero di ossidazione dell’alogeno |
| HCl, KF, NaBr, LiI |
-1 |
| F2, Cl2, Br2, I2 |
Zero |
| HClO, HBrO |
+1 |
| HClO2, Cl2O3 |
+3 |
| HClO3, Cl2O5 |
+5 |
| HClO4, HIO4 |
+7 |
Gli alogeni possono legarsi tra loro detti interalogeni che sono composti molecolari, diamagnetici, ossidanti e spesso instabili ottenuti per reazione degli alogeni stessi:
Br2 + Cl2 → 2 BrCl
Cl2 + 3 F2 → 2 ClF3
Il cloro in ambiente basico dà una reazione di disproporzione con ottenimento di cloruro e ipoclorito:
Cl2 + 2 OH– → Cl– + ClO– + H2O
Se la reazione avviene a caldo si ottiene cloruro e clorato:
3 Cl2 + 6 OH– → 5 Cl– + ClO3– + 3 H2O
In determinate condizioni una miscela di clorato e ipoclorito dà luogo alla formazione di clorito secondo una reazione di comproporzione:
ClO3– + ClO– → 2 ClO2–
Secondo i potenziali standard di riduzione né lo iodio, né il bromo possono ossidare l’acqua ad ossigeno mentre il fluoro e il cloro che hanno potenziali di riduzione maggiori ossidano l’acqua. Il fluoro reagisce con l’acqua secondo la reazione:
2 F2 + 2 H2O → 4 HF + O2
Il cloro reagisce meno vigorosamente secondo la reazione:
Cl2 + 2 H2O → H+ + Cl– + HClO
Gli alogeni reagiscono con i metalli alcalini per produrre sali ionici e solubili in acqua secondo la reazione:
2 M + X2 → 2 MX
Essendo M un metallo alcalino e X2 un alogeno.
Gli alogeni reagiscono con i metalli alcalino terrosi per produrre sali ionici ad eccezione di quelli del berillio secondo la reazione:
Me + X2 → MeX2
Essendo M un metallo alcalino terroso e X2 un alogeno.
Gli alogeni reagiscono con gli elementi del gruppo 13 per formare composti del tipo MX3 dove M è un elemento del gruppo 13. In particolare BF3 è un acido di Lewis e AlX3 assume una struttura dimerica.
Gli alogeni formano composti con gli elementi del gruppo 14 aventi formula generale MX4. Il silicio reagisce con gli alogeni per dare composti del tipo SiX4: a temperatura ambiente SiF4 è un gas incolore, SiCl4 così come SiBr4 sono liquidi incolori mentre SiI4 forma cristalli.
I due metalli presenti nel gruppo 14 formano con gli alogeni sia composti come MeX2 che MeX4.
Gli alogeni formano composti con gli elementi del gruppo 16.
Lo zolfo reagisce direttamente con tutti gli alogeni ad eccezione dello iodio; in particolare lo zolfo si combina spontaneamente con il fluoro per dare l’esafluoruro di zolfo SF6 che è un gas incolore e poco reattivo o il tetrafluoruro di zolfo SF4 che è un gas tossico usato come agente fluorurante molto energico e selettivo.
Dalla reazione tra il tetrafluoruro di zolfo e il tricloruro di boro si ottiene il dicloruro di zolfo che è un liquido rosso utilizzato nella produzione del gas mostarda:
3 SF4 + 4 BCl3 → 4 BF3 + 3 SCl2 + 3 Cl2
Il fluoro si combina con l’ossigeno per dare due composti OF2 e O2F2 in cui l’ossigeno ha numero di ossidazione +1.
Gli alogeni si combinano con l’ossigeno per dare ossidi acidi: in particolare il cloro dà 4 tipi di ossidi: Cl2O, Cl2O3, Cl2O5 e Cl2O7.
Gli alogeni, ad eccezione del fluoro, danno acidi ternari noti come ossiacidi in cui presentano diversi numeri di ossidazione come il cloro che dà HClO, HClO2, HClO3 e HClO4.
Gli alogeni sono inoltre presente in molti composti organici come alogenuri alchilici, alogenuri arilici e alogenuri acilici.